Complicated Presentation and Ultimate Diagnosis of Masked Polycythemia Vera
Presentation
A 51-year-old female presents to the emergency room (ER) with acute abdominal pain. She has a history of lower extremity deep vein thrombosis (DVT) due to presumed antiphospholipid antibody syndrome (APS) and is receiving chronic anticoagulation with warfarin. Physical exam finds the presence of ascites and severe abdominal pain to light palpation. INR the week prior was 2.2, within target range.
Workup
An abdominal CT scan revealed hepatic vein thrombosis (Budd-Chiari syndrome). Complete blood count (CBC) in the ER was unrevealing.
Peripheral blood is sent for BCR/ABL, CALR, MPL, and JAK2V617F mutation. The JAK2V617F mutation returned positive 1 week later, identifying an underlying myeloproliferative neoplasm.
Admission
The patient was started on heparin drip and admitted for a portocaval shunt and liver biopsy. Unfortunately, on post-op day 1, she developed acute bleeding with a drop in hemoglobin from 13 g/dL to 8 g/dL. Partial thromboplastin time was supratherapeutic. The patient was taken back to the operating room for an exploratory laparotomy with hematoma evacuation. Recovery was complicated by ileus, acute kidney injury, and leukocytosis.
Over the next 2 weeks, the patient developed recurrent loculated ascites that required multiple drains placed by interventional radiology . She was treated for a number of infections including enterococcus faecalis and fungal infection of the peritoneal fluid. After 5 weeks of admission, this patient was discharged on low-molecular-weight heparin and warfarin with target INR of 2.5-3.5.
Hematology Referral
Hematology had been consulted during the patient's hospital stay to help with managing anticoagulation. After discharge, she was evaluated in the hematology clinic for a comprehensive consultation of the recent events, history of thrombosis 10 years prior, and newly identified JAK2V617F mutation.
- Repeat testing for APS was negative.
- The patient was found to have iron deficiency, likely, in part, secondary to acute post-surgical blood loss.
- Bone marrow biopsy was performed and revealed hypercellular marrow (50%-70%) with increases in trilineage hematopoiesis and large, atypical megakaryocytes. No increased blasts or evidence of dysplasia. No fibrosis by reticulin staining. Iron stain negative for any stainable iron.
- Myeloid NGS panel: Positive for TET2 and EZH2 in addition to JAK2 already identified
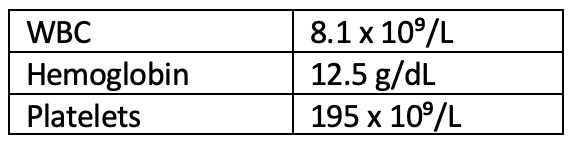
- Repeat CBC without evidence of leukocytosis, erythrocytosis, or thrombocytosis
Discussion
This patient presentation is an example of how difficult it can be to accurately diagnosis and best classify a myeloproliferative neoplasm. Budd-Chiari syndrome can be a presenting sign of underlying polycythemia vera.1 As with this patient, often times, CBC can be normal at the time of presentation with thrombosis. Her WBC and platelet count did increase during the hospitalization; however, this was likely reactive to infectious and bleeding complications post-surgery. Her counts were normal at the time of discharge. The patient’s underlying iron deficiency is likely masking the ability to develop an erythrocytosis.
Careful assessment and discussion with this patient about the need for treatment continues. We have advised starting cytoreductive therapy given the fact that she has developed a thrombotic event even while on therapeutic anticoagulation. A number of treatment options were proposed, and the patient has decided to pursue long-acting interferon. Additionally, she wishes to transition from warfarin to a direct-acting anticoagulant, and given the lack of evidence for APS, this is reasonable. We will continue to monitor her closely and have advised against the use of iron supplementation, and she is considering starting low-dose aspirin for additional prevention.
References
- Lyle L, Kalhagen L. Budd-Chiari Syndrome as Initial Manifestation of Polycythemia Vera: Complexities in the Management of Younger Patients. J Adv Pract Oncol. 2020;11(Suppl 2):10-15. https://doi.org/10.6004/jadpro.2020.11.7.12
