A Young Patient With Abnormal Presentation of Polycythemia Vera
Presentation and Diagnosis
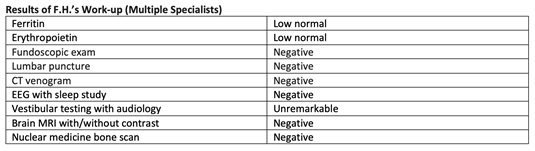
F.H. is an 18-year-old male with no significant past medical history who presented to his primary care provider with new-onset daily headaches. The headaches were associated with photophobia, partial loss of vision, and nausea. In addition to the headaches, F.H. complained of significant fatigue, bone pain, dizziness, and night sweats. He was referred to neurology, where extensive testing was performed after treatment for presumed migraines did not alleviate his symptoms. Laboratory studies were pursued, and his CBC was abnormal, revealing thrombocytosis and erythrocytosis. This table shows the results of his workup, after consulting with several different specialists:
In addition to the other specialists, F.H. also saw pediatric hematology; given his abnormal blood counts, peripheral blood was sent for JAK2 mutation analysis. The results were positive for the JAK2 V617F mutation and the patient was diagnosed with a JAK2+ myeloproliferative neoplasm, not otherwise specified.
On review of his recent history, F.H. had been seen in the emergency department 3 months prior due to chest and flank pain. Workup for the pain had been unremarkable. His CBC at that time also revealed elevated platelets (752,000/μL), Hgb (18.3 g/dL), and hematocrit (54%). His WBCs were normal (7,300/μL).
Interim Monitoring
F.H. started 81 mg ASA daily without a change in symptoms and was then placed on hydroxyurea 1,000 mg/day. Unfortunately, after 3 months he had no change in symptoms and was taken off hydroxyurea. He was then referred from the pediatric clinic to adult hematology for consultation. In the interim, F.H. had been started on antihistamines with some improvement in bone pain, but his fatigue worsened. Review of the labs revealed near normalization of F.H.’s platelet count during treatment with hydroxyurea, but his hematocrit remained >45%. A bone marrow aspiration/biopsy was recommended at that time.
Follow-up
The bone marrow results indicated hypercellular marrow with trilineage proliferation and increased, loosely clustered large-to-giant megakaryocytes; negative for morphologic or immunophenotypic evidence of significantly increased blasts. Iron studies demonstrate scattered iron deposition; ring sideroblasts not recognized; mild focal reticulin fibrosis (MF0-1 of 3). A myeloid next-generation sequencing panel was positive only for JAK2 V617F. Based on his peripheral blood counts, JAK2 positivity, and bone marrow findings, F.H. was classified as having polycythemia vera.
He was seen to review the results, at which time he continued to complain of severe chronic fatigue, night sweats, headaches, and bone pain. After having been off hydroxyurea, his platelet counts had increased again. His CBC was significant for WBC 8,400/μL, Hct 53.7%, and PLT 733,000/μL.
Treatment
Options for disease control were discussed with F.H., including aspirin PLUS phlebotomy alone or in combination with cytoreductive therapy. He opted for a trial of phlebotomies and continued aspirin 81 mg, yet dosing was increased to twice per day. To date, he has had two phlebotomies, and has had a significant improvement in his symptoms. He will continue to be followed closely.
Considerations
Diagnosing an MPN can be very challenging, especially in patients who are younger and in those who present with a constellation of symptoms. Collaboration with multiple specialties may be necessary to rule out alternative etiologies. Critical review of laboratory trends and bone marrow analysis should be performed to arrive at a definitive diagnosis so that appropriate treatment may be pursued.
